SnAP Support and FAQ
Welcome to the SnAP FAQ page!

General Procedure
No special precautions were necessary for reaction set-up and all experiments were performed using identical reaction conditions without substrate-specific optimization.
Step 1: Imine / Ketimine Formation
Imine Formation: To a solution of the SnAP reagent (0.50 mmol, 1.00 equiv) in CH2Cl2 (2.5 mL) at rt was added the corresponding aldehyde (0.50 mmol, 1.00 equiv) and MS 4A powder (ca. 100 mg/mmol). The reaction mixture was stirred at rt for 2–6 h and filtered through a short layer of Celite (CH2Cl2 rinse) to remove the molecular sieves. The filtrate was concentrated under reduced pressure to afford the pure imine. Imine Formation FAQ here!
Ketimine Formation: To a solution of the SnAP reagent (0.50 mmol, 1.00 equiv) in CH2Cl2 (0.5 mL) at rt was added the corresponding ketone (0.50 mmol, 1.00 equiv) and MS 4A powder (ca. 100 mg/mmol). The reaction mixture was stirred at rt for 12 h and filtered through a short layer of Celite (CH2Cl2 rinse) to remove the molecular sieves. The filtrate was concentrated under reduced pressure to afford the pure ketimine. Ketimine Formation FAQ here!
Step 2: Cyclization
Anhydrous Cu(OTf)2 – dried for 2 h at 110°C at 0.1 mbar – (0.50 mmol, 1.00 equiv) is suspended in the appropriate amount of HFIP. 2,6-Lutidine (0.50 mmol, 1.00 equiv) is added and the resulting bluish suspension is stirred at rt for 1 h to afford a dark green suspension. A solution of the imine or ketimine (0.50 mmol, 1.00 equiv) in the appropriate amount of CH2Cl2/HFIP is added in one portion and the resulting mixture is stirred at rt for 16 h. The reaction mixture is diluted with CH2Cl2, treated with a solution of 12% aq NH4OH and brine (1:1), and stirred vigorously for 15 min at rt. The layers are separated and the aqueous layer is extracted with CH2Cl2. The combined organic layers are washed with H2O and brine, dried over anhydrous Na2SO4, filtered, and concentrated. Purification by flash column chromatography affords the corresponding unprotected heterocyclic compounds. Cyclization FAQ here!
Step 3: Purification
The polarity difference of the product heterocycles and tin byproducts allows for simple flash column chromatography to remove tin byproducts. If desired, further purification could be carried out by salt formation of the unprotected N-heterocycles to remove any trace of tin impurities. Purification FAQ here!
Frequently Asked Questions
Imine Formation
Can other solvents be used in case of solubility issues during the imine formation?
Yes! Acetonitrile, benzene, toluene, and 1,2-dichloroethane (DCE) all afforded the imine within the indicated time.
Does leftover aldehyde harm the cyclization if a slight excess was used in the imine formation?
No.
Does leftover SnAP reagent harm the cyclization if a slight excess was used in the imine formation?
No, if Cu(OTf)2 is used in a stoichiometric amount. In case of the catalytic cyclization protocol, the basic primary amine of unreacted SnAP reagents tends to lower the catalyst efficiency resulting in incomplete conversion.
How stable are the resulting imines and can they be purified using flash column chromatography?
As a general rule, imines are stable at room temperature for several hours and no special precautions are needed in the isolation step. Slow decomposition can be observed in CDCl3 if recording of NMR signals takes a prolonged amount of time (more than 1 hour).
Depending on the aldehydes used, few imines can withstand purification using flash column chromatography. However, most imines decompose on silica gel.
Ketimine Formation
Can other solvents be used in case of solubility issues during the ketimine formation?
Yes! Acetonitrile, benzene, toluene, and 1,2-dichloroethane (DCE) all afforded the ketimine within the indicated time.
How can I achieve complete ketimine formation for more challenging substrates?
To a solution of the SnAP reagent (0.50 mmol, 1.00 equiv) in toluene or benzene (1–2 mL) at rt was added the corresponding ketone (0.50 mmol, 1.00 equiv) and MS 4A powder (ca. 100 mg/mmol). The reaction mixture was stirred at 110°C for 12 h in a sealed tube before being cooled to room temperature and filtered through a short layer of Celite (CH2Cl2 rinse) to remove the molecular sieves. The filtrate was concentrated under reduced pressure to afford the pure ketimine.
Does leftover ketone harm the cyclization if a slight excess was used in the imine formation?
No.
Does leftover SnAP reagent harm the cyclization if a slight excess was used in the imine formation?
No, if Cu(OTf)2 is used in a stoichiometric amount.
How stable are the resulting ketimines and can they be purified using flash column chromatography?
As a general rule, ketimines are stable at room temperature for several hours and no special precautions are needed in the isolation step. Slow decomposition can be observed in CDCl3 if recording of NMR signals takes a prolonged amount of time (more than 2–3 hour).
Most ketimines can withstand a short purification using flash column chromatography. However, some ketimines decompose on silica gel.
Cyclization
Is the reaction sensitive to oxygen or water?
No. The reaction is not very sensitive to oxygen or H2O and can be conducted without degassed, extra dry solvents or without pre-dried Cu(OTf)2 with only slightly diminished yields. CaSO4 is not needed to remove trace amounts of H2O in the reaction.
Formation of the Cu(OTf)2 / 2,6-lutidine complex, which solvents can be used?
The pre-mixing of Cu(II) and its ligand can be performed in CH2Cl2, 1,2-dichloroethane (DCE), HFIP, 2,2,2-trifluoroethanol (TFE), and various mixtures containing those solvents.
How to identify protodestannylation in case of low yields and how to prevent this side reaction?
Protodestannylation can be identified in the NMR of the crude reaction mixture. The prododestannylated side products look like the starting imines without the characteristic a-tin protons. Switching from HFIP to less acidic 2,2,2-trifluoroethanol (TFE) reduces the amount of protodestannylated side products. However, longer reaction times are observed using TFE and an increased amount of this protic solvent might be needed to achieve full conversion. In general, an increased amount of the protic solvent (HFIP or TFE) increases the reaction rate, which leads to less protodestannylation.
No reaction is taking place, what should I do?
First, make sure that protodestannylation did not take place. Protodestannylation can be identified in the NMR of the crude reaction mixture. The prododestannylated side products look like the starting imines without the characteristic α-tin protons. Switching from HFIP to less acidic 2,2,2-trifluoroethanol (TFE) reduces the amount of protodestannylated side products. However, longer reaction times are observed using TFE and an increased amount of this protic solvent might be needed to achieve full conversion.
If protodestannylation did not take place and the imine is visible in the crude NMR, an increased amount of HFIP or TFE as the protic solvents can increase the reaction rate. Furthermore, make sure to use Cu(OTf)2 from STREM as material from other suppliers afforded lower yields. Switching from dichloromethane to 1,2-dichloroethane (DCE) and heating the cyclization to 60°C can help to improve conversion for sterically demanding substrates.


Purification
How to identify products for flash column purification?
Product heterocycles are detected by TLC in the crude reaction mixture using both potassium permanganate and ninhydrin stains as UV absorbance might be low in some cases. The product is visible with both developing agents. Using a ninhydrin stain, the products show up as pink-purple spots on TLC. The ligand 2,6-lutidine is visible in UV but does not stain with either KMnO4 or ninhydrin.
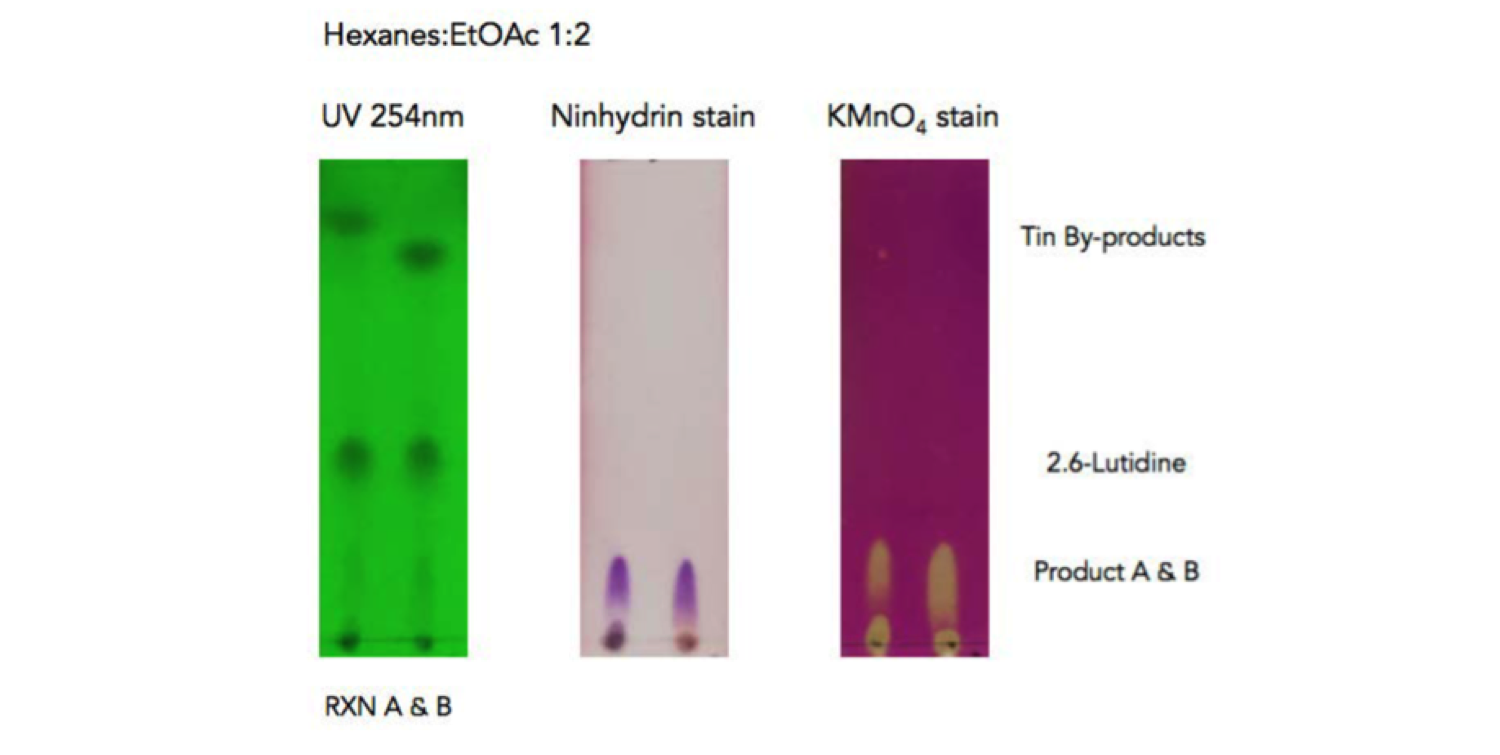
2,6-Lutidine has the same Rf value as the product heterocycles. Can this ligand be separated otherwise?
The boiling point of 2,6-lutidine is 144°C, which is lower than most product heterocycles. Heating the crude reaction mixture to 40°C under reduced pressure (1mbar) removes most of this pyridine derivative before a flash column chromatography is performed.
How to remove tin byproducts most efficiently?
The polarity difference of the product heterocycles and tin byproducts allows for simple flash column chromatography to remove most of the tin compounds.
If desired, most of the tin byproducts can be removed before the flash column chromatography to simplify the purification by column chromatography and afford more pure heterocycles. The crude product is dissolved in CH3CN and washed several times with a small amount of hexanes. The combined hexanes layers are then re-extracted with a small amount of CH3CN. The combined CH3CN layers are concentrated under reduced pressure to afford the crude product with much less tin residues compared to the original one (Berge, J. M.; Roberts, S. M. Synthesis 1979, 471–472).
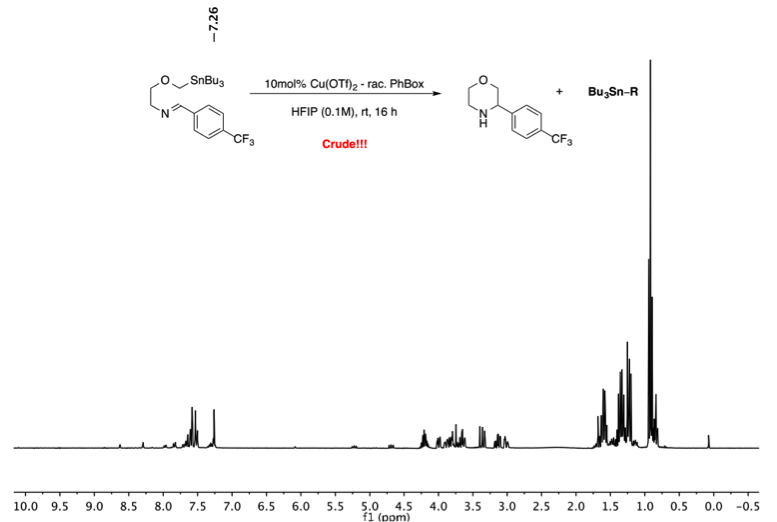
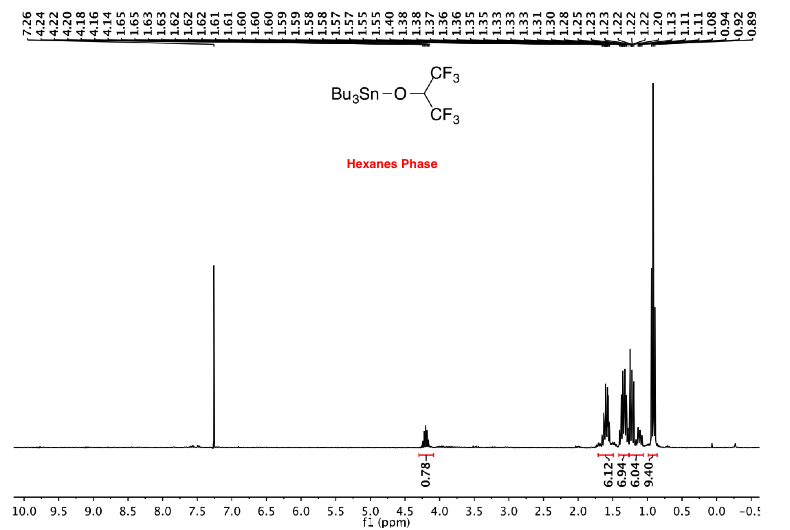
If desired, further purification could be carried out by salt formation of the unprotected nitrogen heterocycles to remove any trace of tin impurities.
Common methods such as extraction using aqueous KF or column chromatography using silica gel mixed with either KF or K2CO3 are not suitable to remove those impurities due to the nature of the tin impurities (Bu3SnOR).
If you have any further questions, please don't hesitate to contact us!